



Enveda’s PRISM (Pretrained Representations Informed by Spectral Masking) model was trained on 1.2 billion small molecule mass spectra, the largest training set of small molecule mass spectra ever assembled.
 01
01One of the most influential advances in machine learning during the last decade has been the advent of large pretrained foundation models like Google’s BERT for language understanding or OpenAI’s GPT family for language generation. These models were trained on massive datasets of unlabeled data using a technique called self-supervised learning. This is in contrast to most previous methods, which relied on large databases of labeled data (e.g., thousands of pictures of cats with the label “cat”). The ability of machine learning models to make connections and identify patterns without being explicitly taught them is powerful because there is far, far more unlabeled data than labeled data in the world.
Typically, self-supervised learning includes a process of masking, which involves hiding parts of the data and then having the model predict the missing piece given the context that remains (more on this later). The ability to learn the meaning of language by ingesting raw text without the need for explicit human annotations is what allowed these models to scale to billions of examples and more accurate outputs. They are called foundation models because once pre-trained on large unlabeled data, these foundation models generalize to many different tasks, often by being fine-tuned on smaller labeled datasets.
The goal of any foundation model is to increase the accuracy of downstream predictions, and we built PRISM with the goal of further improving the models that underlie our novel drug discovery platform. In this blog post, I will discuss the datasets that we used, our training methodologies, the improvements we saw after integrating PRISM into our models, and what’s next.
02Living things are full of small organic molecules that are products of metabolism (called metabolites). These molecules serve a huge range of critical functions, including mediating cell signaling, storing and converting energy, building cellular structures and tissues, defending against pathogens, and much more. The study of metabolite structures and their respective biological functions is called metabolomics.
One of the fundamental methods for determining which metabolites exist in a given living organism (such as a blade of grass) is liquid chromatography and tandem mass spectrometry, or LC-MS/MS. In LC-MS/MS, samples are separated out along a gradient, then individual molecules are fragmented to pieces, and the masses of the separate fragments are measured and collected into an MS/MS mass spectrum.
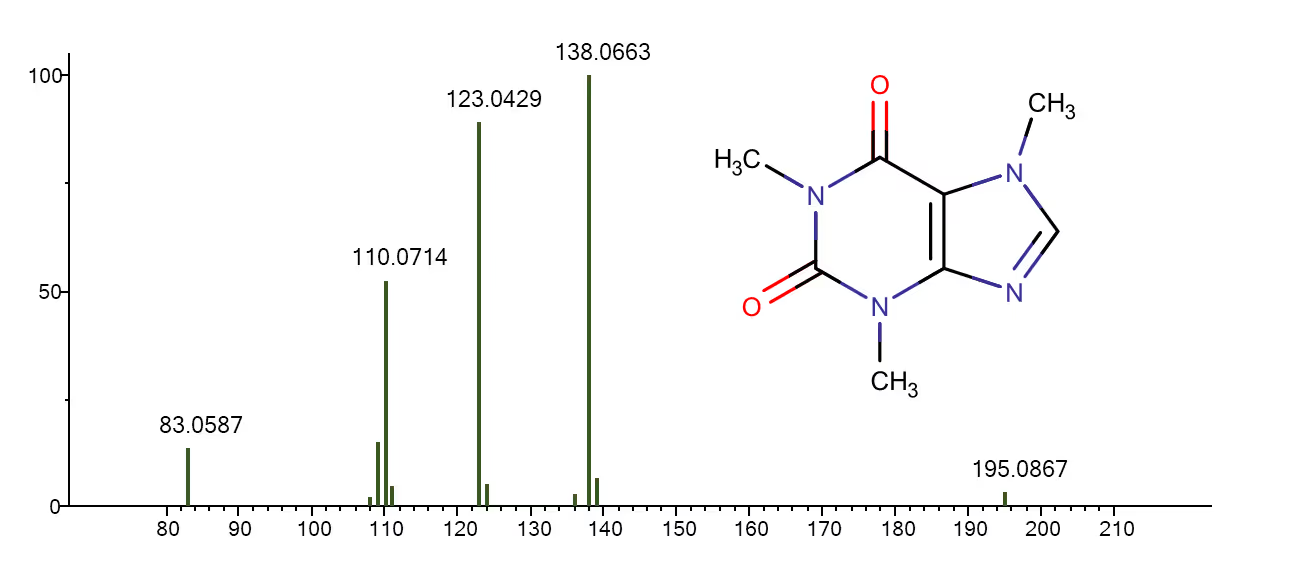
Tandem mass spectrum (MS/MS) of caffeine acquired on a quadrupole time-of-flight (Q-TOF) mass spectrometer. The spectrum displays peaks that correspond to different parts of the molecule, which are formed when the ions are fragmented through collision-induced dissociation (CID) breakages (source: MoNA).
Mass spectrometry data does include structural information on the original molecule, and this information can be used to identify molecules and structures – if that molecule is “known” and has a reference spectrum to compare to. But, shockingly, in any given biological sample (taken from, say, human blood, plant extracts, or soil samples) only a few percent of molecules are likely to be “known” and thus identifiable. This is because the vast majority of the small molecules that exist in nature have never been identified or characterized by scientists before.
Our goal is to use ML to be able to interpret MS data so that we can predict the structures and identify molecules that have never been seen by science before, as we believe the 99.9% of unknown natural molecules represents the largest untapped resource for new medicines on the planet. We’ve previously discussed Enveda’s ML approaches for interpreting mass spectra in previous posts (MS2Mol, MS2Prop and GRAFF-MS).
Machine learning typically requires a lot of data, and in metabolomics there is a lot more unannotated experimental data than annotated data, precisely because most molecules are unknown. In order to annotate an MS/MS spectrum, you must positively determine the structure of the molecule being profiled, usually by first isolating the molecule or by synthesizing it, both of which are time consuming and expensive experiments. But a single experimental profiling of a biological sample can generate thousands of unannotated mass spectra corresponding to the thousands of (mostly unidentified) molecules present in those samples.
You can see this difference reflected in repository sizes: publicly available annotated spectra in repositories like NIST or MoNA contain on the order of a few million mass spectra between them, generated from no more than 60,000 or so unique molecules. In contrast, repositories of raw unannotated data like MetaboLights (Haug et al., 2020), the Metabolomics Workbench (Sud et al., 2016) and GNPS (Wang et al., 2016) each contain hundreds of millions of unique mass spectra, and likely represent a much broader sampling of the potentially billions of distinct small molecules hypothesized to exist in nature (Reymond, 2015).
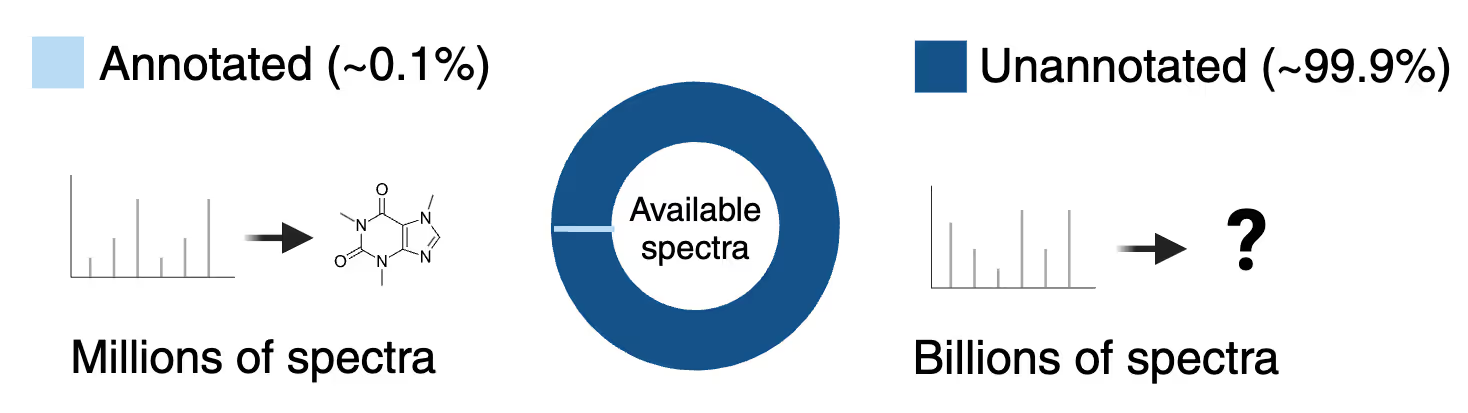
Most machine learning models use annotated mass spectra. However annotated data represents only a tiny fraction of total MS/MS spectra available for training.
03Despite a wealth of unlabeled data, machine learning models have historically used labeled experimental spectra for mass spectral interpretation. This includes tasks like predicting molecular properties (as MS2Prop), looking up similar molecules from spectral references (as MS2DeepScore), predicting structures from sets of candidate spectra (as CSI:FingerID or MIST), or de novo generation of structures directly (as MS2Mol or MSNovelist). While the vast majority of machine learning is done on labeled data, training representations on unlabeled data has begun to be explored, with ever increasing size of data sets and complexity of algorithms. Early versions included using topic modeling and word embedding algorithms, typically trained on tens of thousands of spectra. In recent weeks the first modern transformer architectures trained on larger datasets have emerged, including the models GleaMS and LSM1-MS2, both of which are self-supervised transformer models trained on 40M and 100M tandem mass spectra, respectively.
At Enveda, we have trained a foundation model at the scale of the entire discipline, training on virtually all publicly available quality spectra combined with an equal number of proprietary spectra gathered on Enveda’s high-throughput profiling platform. Our model is called PRISM (Pretrained Representations Informed by Spectral Masking), and at 1.2 billion training spectra and 85 billion tokens, PRISM has seen over an order of magnitude more data than any previous mass spectrum model (for comparison, GPT-3 was originally trained on around 300 billion tokens). It improves performance on a wide variety of prediction tasks, and crucially we see that performance continues to increase with the size of the training set without leveling off.
Let’s dive into the details!
04Enveda’s PRISM foundation model is a self-supervised learning model with an architecture adapted from the popular Bidirectional Encoder Representations from Transformers (BERT) family of natural language encoders, but adapted for use with tandem mass spectra. BERT uses a training framework called Masked Language Modeling (MLM). The basic idea of MLM is that you can teach a model the structure of natural language by “masking” one or more pieces of the sentence and asking the model to predict the missing pieces given the context from the others. For example, given the sentence: “The dog tried to follow the person”, we could train the model by masking, or hiding, the word “follow”, so the model sees “The dog tried to [MASK] the person.” Then we ask the model to use the context provided by the other words to predict the masked word.
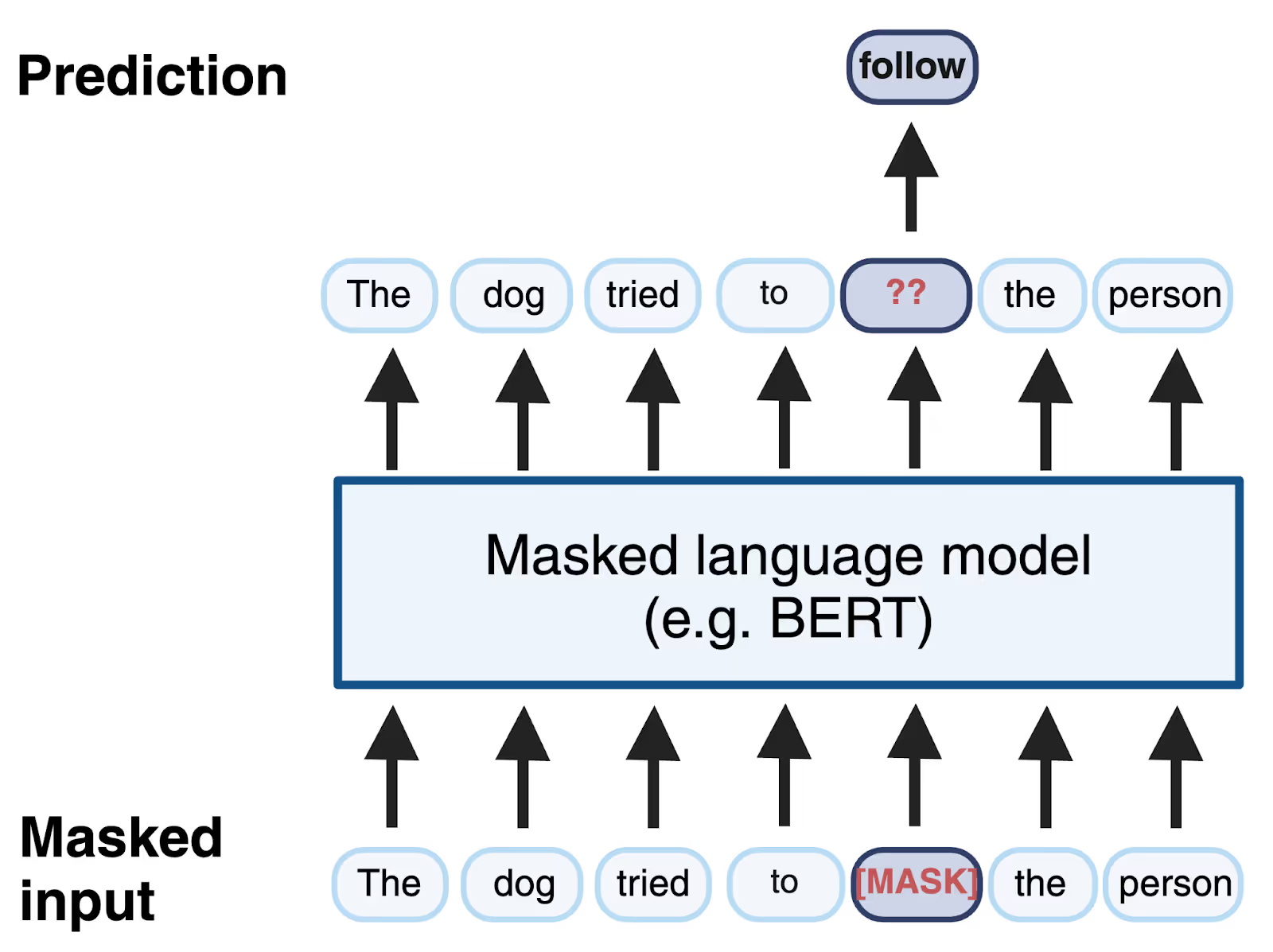
Masked language modeling, a form of self-supervised learning where tokens of an input sentence are hidden from the model and the model is trained to predict the identity of the missing tokens by using the context of the rest of the sequence.
Across many such examples, if the model learns to predict that the missing word is something like “FOLLOW”, and if it can do this consistently across many kinds of sentences, then in some sense the model has learned the grammar and structure of the English language without explicitly being told anything about the meanings of the words or sentences. And since all it needs for masked language modeling is raw text, it can be trained on many billions of such examples. Frameworks of this sort are the basis for the foundational large language models (LLMs) that dominate the artificial intelligence world today.
At Enveda we use a similar masking protocol for the PRISM foundation model, only masking masses instead of words. We call this Masked Peak Modeling. In this approach, for each example mass spectrum – which consists of the masses and intensities that result from breaking apart a molecule and that are represented as “peaks” on the MS2 – we randomly mask the masses of 20% of the peaks. The model’s job is to predict the mass of the missing peaks given the context of the peaks that remain.
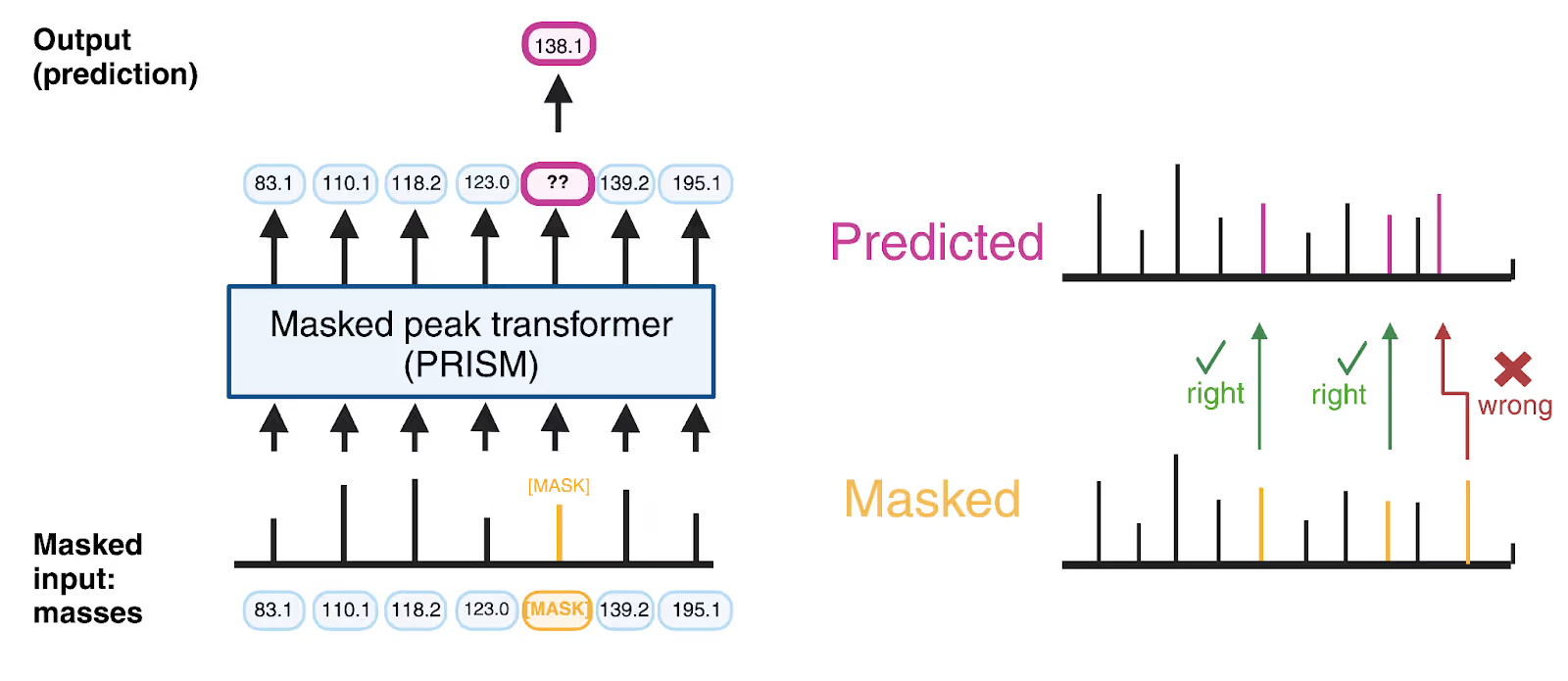
PRISM learns the grammar of unannotated mass spectra by predicting the masses of masked fragment peaks.
To train PRISM, we gathered the largest small molecule MS/MS training set to date, consisting of 1.2 billion high quality small molecule MS/MS spectra. About half of those spectra (around 600 million) came from combining all three of the major public data repositories mentioned earlier (GNPS, MetaboLights, Metabolomics Workbench), while the other 600 million spectra came from Enveda’s internal metabolomics platform, which we use to search for new potential medicines from nature.
As with natural language, PRISM is able to leverage the billions of unannotated spectra to learn the “grammar” of chemistry. We find that the internal representations, or embeddings, of spectra that PRISM learns in this process end up encoding facts about the structures and properties of the underlying molecules without ever being taught them, grouping classes of molecules together and even accounting for experimental differences like machine type and collision energy.
05Of course, being able to guess missing masses is not really the end goal of a foundation model. The hope of PRISM is that a model that has learned from billions of unannotated spectra will better “understand” new spectra of unknown molecules from the (proverbial and literal) wild, and will be better at interpreting these spectra in order to predict the properties and structures of the underlying molecules – information that is critical to the drug hunters on our team.
To illustrate this point we examined the task of predicting a suite of chemical properties of molecules solely from their mass spectra. We predicted properties like the reactiveness, synthetic accessibility, and numbers of different kinds of rings and atoms, all of which are important for discovering new medicines from nature. We compared two different models. The first, which we will call “Standard,” was a typical machine learning model trained on around 2 million annotated spectra from around 60,000 reference molecules. The second, “Standard + PRISM” is the same model architecture, but initialized using PRISM’s pretrained weights before being fine-tuned on the labeled data. This comparison allowed us to attribute any difference in prediction performance to just the effect of the PRISM pretraining.
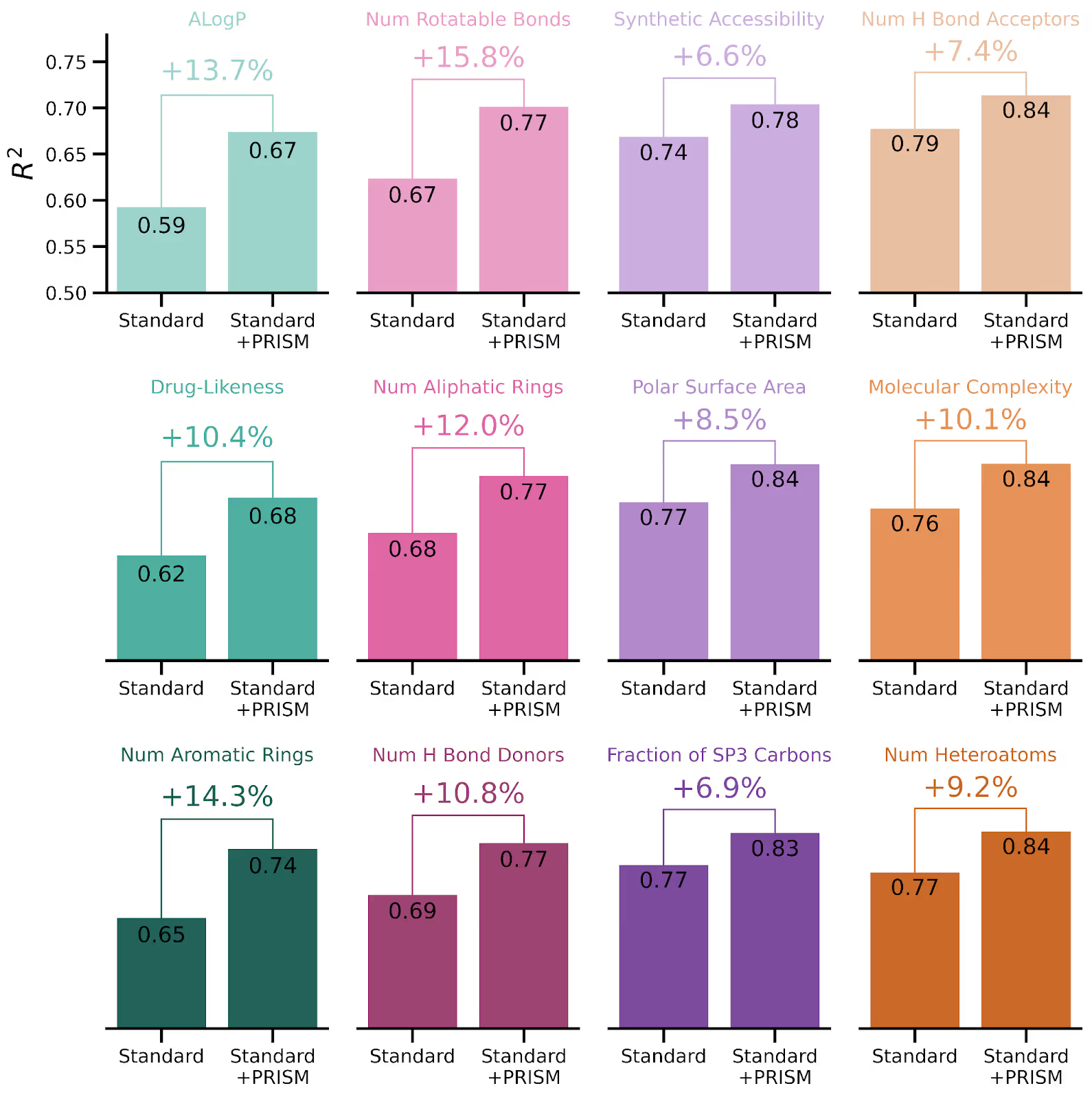
PRISM helps predict relevant drug-likeness properties of molecules from their mass spectra. Here we show 12 different chemical properties predicted from spectra, either by a standard machine learning model (MS2Prop) trained only on labeled data (left bars) or the same model trained on the same labeled data but initialized with PRISM pretraining (right). PRISM results in a relative increase in R^2 between actual and predicted values of 7% and 16%)
Across all properties we predicted, PRISM provided a notable improvement to the predictions compared to versions of the same models trained on the same data but without PRISM. To validate that this success was not limited to property prediction, we also tested the ability of PRISM to match unknown spectra to the closest match in a spectral reference library, and achieved a 23% relative improvement on that task when compared to a standard ML model trained for that purpose but without PRISM pretraining.
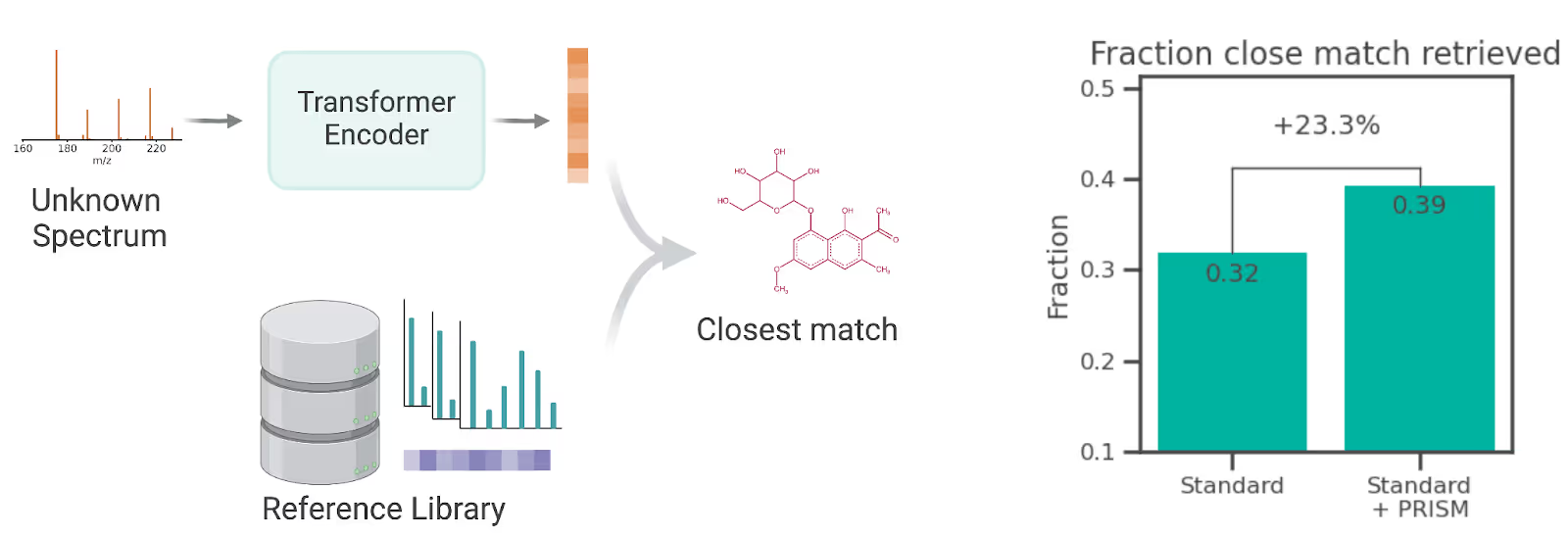
PRISM also improves the task of predicting structures for unknown spectra by finding the closest match in a spectral reference library. Here we train a standard contrastive learning representation model (either with or without PRISM pretraining) and then use it to retrieve the closest molecule from a spectral reference library. With PRISM, we retrieved a close match for 23.3% more test spectra than without PRISM.
Better predictions translate into faster drug discovery as Enveda’s drug hunters sift through hundreds of such molecule predictions, looking for those with the right properties to become a drug.
06Enveda’s PRISM foundation model represents the largest small molecule mass spectrometry machine learning model to date. However, a billion spectra is only the beginning. Enveda’s automated pipeline for mass spectral profiling has already generated hundreds of millions of MS/MS spectra, and as we scale our search for new medicines, so too will we scale the size and diversity of our experimental data. As we’ve seen, large amounts of raw data for training equals better predictive models, and better models enable us to decode the chemistry of nature to find interesting new medicines.
Stay tuned for more details on PRISM, our other ML x Mass Spec work, and platform technology updates in the coming months.